

AnalyzeAlign Explanation
Purpose
AnalyzeAlign will perform several analyses on any alignment.
- Show Weblogos.
- Calculate frequency by position.
- Find variants within the alignment.
LANL alignment type
We create the following different regional subalignments:
- the “FULL” alignment represents the coding regions, from the 5’-most start-codon (orf1ab) to the 3’-most stop-codon (ORF10), NC_045512 bases 266-29,674
- “NEARCOMP”, near-complete, includes the most-commonly-sequenced region of the genome, bases 55-29,836
- "COMPLETE" includes sequences matching the NC_045512 sequence from start up to the poly-A tail, bases 1-29,870
- "Regional COMPLETE" is available for Spike, ORF3a, nsp3, and RdRp and includes sequences covering the region. For example, the Spike COMPLETE alignment includes sequences matching the NC_045512 bases 21,563-25,384
Position/range to analyze
You must indicate what part of the alignment to analyze.
- Maximum of 30 positions!
- Enter coordinate values. These values must be based on residues of NC_045512.
- Comma delimit multiple values.
- The value "end" is accepted.
- Example input: 1, 10, 20-29, 450-end
Group results
The default is to analyze all sequences together as a single group. However, your sequences may contain groups that you want to analyze separately by date or region.
Combine logos into a page: allows you to download all your logo images in a single PDF file. Specify EITHER the length OR width of the display matrix, and its orientation. The other matrix number (length or width) will be determined by the script, based on the actual number of groups in the output. If you aren't sure how to use these options, just keep the default concatenation (1 x __) and orientation; this will always work.
- Include removed logos: if checked, will show the logos of removed characters in the page where all logos are presented.
Logo options
The WebLogos in this tool are based on a slightly modified Weblogo 3. For details about options, see WebLogo 3 User's Manual.
Logo size
Max number of logo stacks per line
"Stacks" refers to the vertical columns of the logo. Long logos may be broken by line breaks, and this option allows you to choose the number of positions per line.
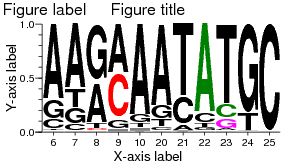
Figure title
See illustration at right.
Figure label
See illustration at right.
X-axis label and stack annotations
By default, the columns are labeled by the position numbers you chose. You can override these numbers by providing a comma-separated list of characters to replace the default.
Y-axis units
Choose the units for the Y-axis.
Y-axis label
By default, the y-axis will be labeled based on your choice of y-axis units. You can override this label with your own.
Color schemes for WebLogo
The WebLogo software provides several predefined options for how to color the residues in your logos. Instead of using the standard options, you can choose colors:
Paste or upload custom color scheme.
Use this option to specify a highlight color for a few specific residues. In the image above, the C at position 8 and T/C at position 9 were colored red (#FF0000 in hexadecimal format, see below), by inserting this custom color scheme:
#FF0000: 8 C 9 TC Green: 22 A 23 C Magenta: 23 G
You can define the colors using
- HTML color names
- a RGB hex triplet which is a six-digit number, where each pair of digits is a hexadecimal number and represents the red, green and blue components of the color. Hexadecimal color codes may be specified with notation using a leading number sign (#).
Specify symbol colors.
Input custom colors for any specific symbols. Colors can be defined by clicking the color text box to display a color palette and choosing one or typing a RGB triplet as explained in the paragraph above.
Remove symbols from logo
This option allows you to change the appearance of the sequence logo image(s).
show all
If the default 'show all' is chosen, the logo is presented as usual, showing the abundance of each residue in the alignment. At right, the epitope RPNNNTRKSI was aligned to the LANL HIV-1 filtered web alignment.
remove: consensus of alignment
If this option is chosen, the images omit the most common residue at each position of the alignment. At right, the epitope RPNNNTRKSI was aligned to the LANL HIV-1 filtered web alignment.
remove: consensus of seq group
This option is similar, but removes consensus of each sequence group, rather than the consensus of the whole alignment.
remove: residues of 1st sequence
If this option is chosen, the images omit the residue that occurs in the first sequence of the alignment. For the LANL database alignments, the first sequence is always the reference sequence. For user alignments, the first sequence might be a vaccine sequence, for example, being compared to the alignment it was derived from. At right, the epitope RPNNNTRKSI was aligned to the LANL HIV-1 filtered web alignment.
remove: residues of 1st seq of seq group
This option is similar, but removes the residues of the first sequence of each sequence group, rather than the first sequence of the whole alignment.
remove: user-specified
Remove residues corresponding to a user-specified sequence. Enter a single sequence in raw format (no sequence name). This sequence must have exactly the same number of residues as defined in "Positions/range to analyze".
Delete Gaps and Shift
If the "Delete gaps and shift" option is selected, then gaps placed to bring sequences into alignment will be squeezed out and the alignment shifted rightwards (toward the C-terminal end). For example, suppose your query has a one-amino acid insertion relative to most other sequences, then following alignment:
QUERY VARELHP REF VAR-LHP seq2 VAR-LHP seq3 VAR-LFP seq4 VAR-LMP
would be presented like this with gaps deleted:
QUERY VARELHP REF QVARLHP seq2 QVARLHP seq3 QVARLFP seq4 QVARLMP
Q is the amino acid one position to the left of the V. As a result of squeezing gaps and shifting characters rightward, alignments in gappy regions will look "bad."
NOTE: The delete gaps option is useful for aligning immunologically reactive epitopes, because in such cases it is particularly important to maintain the alignment of the C-terminal anchor residues.
Mark potential N-linked glycosylation sites
When 'yes' is chosen, any asparagine (N) occurring in the pattern NxS or NxT (x = any amino acid except proline) will appear as "O". For more information about N-linked glycosylation, see N-GlycoSite.
Cut-off for calculating frequency by position
The tool will calculate the frequency of each nucleotide or amino acid at each position. The frequency table will show only the residues with the highest representation(s), as determined by a cutoff. If the cutoff is 100%, all residue frequencies will be shown. If the cutoff is 95%, the most frequent residues will be shown, up to a cumulative total of 95%, then all others will be presented as "other". Lumping together the infrequent residues can be a useful simplification, particularly for protein sequences.
Master sequence for finding variants
Variants are defined with one "master" sequence used as the basis of comparison. You can choose which sequence will be the master.
To choose a "user-selected" sequence, enter a single sequence in raw format (no sequence name). This sequence must have exactly the same number of residues as defined in "Positions/range to analyze".
This option does not affect the logo or the calculation of frequency by position.
Additional Resources
- LANL Alignments describes the different types of premade alignments available.
- WebLogo 3 User's Manual
Questions or comments? Contact us at seq-info@lanl.gov.